Topic 3.4. Spasticity and spasms in Friedreich ataxia
This chapter of the Clinical Management Guidelines for Friedreich Ataxia and the recommendations and best practice statements contained herein were endorsed by the authors and the Friedreich Ataxia Guidelines Panel in 2022.
Topic Contents
3.4.1 Spasticity and spasms in Friedreich ataxia
3.4.1.1 Prevalence of spasticity in Friedreich ataxia
3.4.1.2 Spasticity in late-onset Friedreich ataxia (LOFA)
3.4.1.3 Spasticity in Friedreich ataxia with retained lower limb tendon reflexes (FARR)
3.4.1.4 Spasticity in Acadian FRDA/Acadian spastic ataxia
3.4.1.5 Spasticity in compound heterozygotes with FXN point mutations
3.4.2 Functional consequences of spasticity and spasms
3.4.3 Management of spasticity and spasms
3.4.3.1 Pharmacological treatments
3.4.3.2 Physical Therapy
3.4.3.3 Surgical Treatment
Disclaimer / Intended Use / Funding
Disclaimer
The Clinical Management Guidelines for Friedreich ataxia (‘Guidelines’) are protected by copyright owned by the authors who contributed to their development or said authors’ assignees.
These Guidelines are systematically developed evidence statements incorporating data from a comprehensive literature review of the most recent studies available (up to the Guidelines submission date) and reviewed according to the Grading of Recommendations, Assessment Development and Evaluations (GRADE) framework © The Grade Working Group.
Guidelines users must seek out the most recent information that might supersede the diagnostic and treatment recommendations contained within these Guidelines and consider local variations in clinical settings, funding and resources that may impact on the implementation of the recommendations set out in these Guidelines.
The authors of these Guidelines disclaim all liability for the accuracy or completeness of the Guidelines, and disclaim all warranties, express or implied to their incorrect use.
Intended Use
These Guidelines are made available as general information only and do not constitute medical advice. These Guidelines are intended to assist qualified healthcare professionals make informed treatment decisions about the care of individuals with Friedreich ataxia. They are not intended as a sole source of guidance in managing issues related to Friedreich ataxia. Rather, they are designed to assist clinicians by providing an evidence-based framework for decision-making.
These Guidelines are not intended to replace clinical judgment and other approaches to diagnosing and managing problems associated with Friedreich ataxia which may be appropriate in specific circumstances. Ultimately, healthcare professionals must make their own treatment decisions on a case-by-case basis, after consultation with their patients, using their clinical judgment, knowledge and expertise.
Guidelines users must not edit or modify the Guidelines in any way – including removing any branding, acknowledgement, authorship or copyright notice.
Funding
The authors of this document gratefully acknowledge the support of the Friedreich Ataxia Research Alliance (FARA). The views and opinions expressed in the Guidelines are solely those of the authors and do not necessarily reflect the official policy or position of FARA.
3.4 Spasticity and spasms in Friedreich ataxia
Michael H. Parkinson, Sarah C. Milne, Paola Giunti and Sub H. Subramony
This chapter describes spasticity and spasms associated with Friedreich ataxia, the functional consequences of spasticity and spasms, and strategies for managing spasticity and spasms. In making recommendations for management, the authors were tasked with answering the question:
For individuals with Friedreich ataxia, what management strategies could be implemented for spasticity and spasms?
3.4.1 Spasticity and spasms in Friedreich ataxia
Spasticity is the presence of increased muscle tone (hypertonia) caused by a lesion of the upper motor neurons (or pyramidal tracts). Spasticity is distinguished from rigidity because of its velocity-dependent nature and absence of associated extra-pyramidal features. It can cause muscular stiffness, spasms and pain.
Friedreich ataxia (FRDA) can be associated with increased levels of spasticity and muscular spasms and cramps, particularly in the late-onset forms of the disease and after long disease duration. Spasticity can affect many parts of the body and contribute to many of the problems experienced by individuals with FRDA, such as dysarthria and dysphagia, and deficits in the upper and lower limbs, such as problems with manual dexterity and gait. In FRDA, because of the coexisting pathological involvement of the dorsal root ganglia and the cerebellum, the exact contribution of spasticity and upper motor neuron (UMN) pathology to the motor dysfunction may be difficult to assess. In addition, involvement of the sensory pathways may mask some of the traditional signs associated with spasticity, such as hyperreflexia.
3.4.1.1 Prevalence of spasticity in Friedreich ataxia
There are a number of publications that report the prevalence of spasticity or hypertonia in individuals with FRDA, which ranges between 12% and 15% in individuals with typical onset FRDA and 33% and 40% in individuals with late onset FRDA (LOFA) (1-4).
Geoffroy and colleagues (3) described 33 patients with ‘typical’ FRDA of whom 15.1% had spasticity. Harding (4) described 115 patients with FRDA from 90 families in the UK. She found 12.2% had increased lower limb muscle tone but none of the patients had increased upper limb tone. Finger contractures were found in 11.3%. Montermini and colleagues (5) studied the phenotypic variability in 100 patients with FRDA, including eight patients with LOFA, six patients with FRDA with retained reflexes (FARR) and 44 patients with Acadian FRDA. Although they did not comment on the prevalence of spasticity in the main cohort, the Acadian group included four patients who had spasticity.
Badhwar and colleagues (6) reviewed the literature describing people with FRDA presenting with spasticity. They identified 21 patients: 38% had one GAA expansion of fewer than 200 repeats; 33% had a point mutation (either G130V, R165C or R165P); and 10% were homozygous for GAA expansions of greater than 200 repeats. They also described a family in which one individual, homozygous for a 750 GAA repeat expansion, had marked spasticity, but other individuals with similar or smaller expansions had different presentations without spasticity. They concluded that individuals with such ‘mid-size’ expansions can develop spasticity and that disease course is difficult to predict from expansion size as measured from peripheral blood samples.
Milne and colleagues (7) examined the presence of spasticity in the gastrocnemius and soleus muscles, using the Modified Tardieu Scale (MTS), in 31 individuals with FRDA. All participants had spasticity in at least one of the muscles examined, and 39% of ambulant and 69% of non-ambulant participants had contracture in one or both of their gastrocnemius muscles.
Corben and colleagues (8) similarly used the MTS to examine the presence of spasticity in the upper limbs of 15 individuals with FRDA. Spasticity in wrist and finger flexors was found in 68% of participants and 37% had contracture in at least one joint in their hands. The severity of spasticity was significantly inversely correlated with age at disease onset.
It should be noted that most studies, except those by Harding (4) and Corben and colleagues (8), report spasticity affecting lower limbs. Clinically, it is apparent that some individuals with FRDA may also present with spasticity affecting upper limbs, warranting further examination.
3.4.1.2 Spasticity in late-onset Friedreich ataxia (LOFA)
Bhidayasiri and colleagues (2) studied 13 patients with LOFA (defined as having clinical onset of symptoms after the age of 25) and compared them with 13 patients with typical age of onset matched for sex and clinical severity. Patients with LOFA had significantly smaller GAA repeat size than those with typical FRDA (176±135 vs 490±52 for the smaller allele). Lower limb spasticity, retained reflexes, lack of sphincter disturbance and normal echocardiogram were significantly more common in the LOFA group. Klockgether and colleagues (1) reported similar results with spasticity present in 15% of individuals with typical onset FRDA and 33% in those with LOFA. Furthermore, Ragno and colleagues (9) described lower limb spasticity in an individual with LOFA while Gates and colleagues (10) described spastic ataxia in three individuals with LOFA.
3.4.1.3 Spasticity in Friedreich ataxia with retained lower limb tendon reflexes (FARR)
Coppola and colleagues (11) studied 101 patients who were diagnosed with a homozygous FXN GAA repeat expansion. Of these, 11 individuals (10.9%) had retained tendon reflexes in the lower limbs and were described as having the clinical phenotype of FARR. They varied in age at onset of symptoms from 13 to 45 years. The mean size of the smaller GAA expansion was 408, varying between 120 and 784. Of the 11 individuals with FARR, four (36.4%) had increased lower limb tone.
3.4.1.4 Spasticity in Acadian FRDA/Acadian spastic ataxia
Richter and colleagues (12) studied a milder, more slowly progressive variant of FRDA in 10 Acadian families from New Brunswick, Canada. A recessive ataxia affecting Acadians from the Maritime Provinces of Canada had already been described, which for historical reasons is linked with a cluster in the Cajun populations of Louisiana, USA (13). Three individuals from two of these families had spastic ataxia.
3.4.1.5 Spasticity in compound heterozygotes with FXN point mutations
Gellera and colleagues (14) in their series of 13 compound heterozygotes with one GAA repeat expansion and one of seven point mutations in the FXN gene, found mild gait spasticity in one individual with a c.157delC (p.R53AfsX75) mutation. Cossée and colleagues (15) compared the clinical features of 25 such compound heterozygotes from 19 families with 196 individuals homozygous for the GAA repeat expansion. Those with truncating mutations, or missense mutations in sequences encoding the carboxy-terminal half of the frataxin protein had a similar phenotype to those with a homozygous GAA expansion. However, individuals with a missense mutation in sequences encoding the amino-terminal half of the frataxin protein (D122Y and G130V) had a milder clinical phenotype with early-onset spastic gait. In addition, these people often had retained upper and lower limb reflexes, no dysarthria, and little or no ataxia.
Refer to Chapter 12 for further discussion of point mutations/deletions.
It seems therefore that individuals with a relatively short GAA expansion in the FXN gene (<200 repeats) commonly have a later age at onset of symptoms and often have an atypical presentation which may mimic progressive spastic paraparesis. Such individuals should be monitored for the development of spasticity, and appropriate interventions put in place. It therefore follows that patients who present with some features of FRDA but who have atypical features such as late onset or retained reflexes, or who present with spastic paraparesis for which no other cause is found, may warrant genetic testing for FRDA, which may include testing for compound heterozygous point mutations if heterozygosity for a GAA expansion is found. However, given the considerable phenotypic variability of FRDA, it is highly likely spasticity will still develop in individuals with larger GAA expansions.
3.4.2 Functional consequences of spasticity and spasms
In clinical practice, it is observed that lower limb spasticity and abnormal posturing can be a common and distressing feature of FRDA. Spasticity involves an increased level of muscle tone causing stiffness, tightness, and sometimes pain and discomfort. This prevents normal limb movement and can impact on normal walking pattern and motor function (7). Spasms are a form of spasticity where the muscle forcibly contracts spontaneously or when undertaking activity, and can cause difficulty in the use of limbs as well as pain and discomfort. Individuals who do not demonstrate spasticity overtly on examination may well have brisk reflexes once chair-bound and are often bothered by severe flexor spasms, a manifestation of the UMN syndrome. This can severely affect sleep and quality of life.
In the late stages of the disease, people with FRDA often develop talipes equinovarus causing difficulties with standing, transfers and wheelchair positioning, leading to progressive disability and increased carer need.
Upper limb spasticity, in combination with upper limb muscle weakness, also has a detrimental impact on the ability to complete daily activities (8, 16). Corben and colleagues (8, 16) found an unusual pattern of upper limb spasticity that predominantly affects wrist and finger flexion in individuals with FRDA.
3.4.3 Management of spasticity and spasms
Few trials of interventions for spasticity in individuals with FRDA have been undertaken. Indeed, there is only one case history which described this subject specifically (17). The underlying neurophysiological mechanisms of spasticity and spasms are likely to be similar in FRDA to those in more common conditions, such as stroke and multiple sclerosis, in which spasticity is a more prominent feature and for which there is a greater evidence base. Decisions regarding spasticity management in FRDA therefore need to be made pragmatically on the basis of evidence from other conditions and clinical experience. Comprehensive reviews of the management of spasticity in multiple sclerosis (MS) are given in the 2014 NICE Clinical Guidelines (No. 186) undertaken by the UK’s National Institute for Health and Care Excellence (18) and the Cochrane Library of systematic reviews (19, 20). Cochrane reviews also exist for spasticity associated with spinal cord injury (21), amyotrophic lateral sclerosis (22) and cerebral palsy (23, 24).
The purpose of treating spasticity is to maintain mobility, capacity to stand and transfer, and upper limb function; to reduce distressing symptoms of pain and spasms, in particular to reduce nocturnal spasms and so improve sleep and prevent fatigue; to facilitate seating, transferring, toileting or dressing and thereby reduce caregiver burden or reliance, and promote independence; to prevent contractures and thereby reduce the development of long-term disability; and to prevent the development of pressure sores or skin ulceration and so reduce resulting comorbidity.
3.4.3.1 Pharmacological treatments
Medications should be used in combination with patient education and physical therapy. The evidence base for all agents is weak with very few placebo-controlled trials conducted. Oral medications or intrathecal baclofen may work best for generalized spasticity, whilst intramuscular botulinum toxin or phenol injections can help with more focal problems. In all cases, there can be paradoxical worsening of mobility or functioning, such as in transferring, by reducing spasticity and unmasking weakness (25). We found no evidence of specific trials of oral baclofen, tizanidine, benzodiazepines, dantrolene sodium, gabapentin, cannabis extract or quinine sulphate in FRDA.
Oral Medications
Baclofen
The most commonly used agent is baclofen. Clinical trials of baclofen have included patients with stroke, cerebral palsy, spinal cord injury and MS. Although most have shown reduced levels of hypertonia and spasms, few have shown improved levels of functioning. Baclofen should be started at a dose of 5-10 mg per day, generally at night due to its sedating side effects. The dose can be increased gradually in three divided doses to a maximum of 100-120 mg per day. Side effects include drowsiness, confusion and dizziness. It should be used with caution in the presence of epilepsy as it lowers seizure threshold. Abrupt withdrawal should be avoided as this can cause seizures, anxiety, confusion and hallucinations (26, 27).
Tizanidine
Placebo-controlled trials in stroke, MS and spinal cord injury have shown reduced muscle tone, spasms and clonus, but again have not demonstrated functional benefit. Tizanidine causes drowsiness, dizziness, dry mouth, hypotension, hallucinations but also impairment of liver function tests. It is advised to check these monthly for the first four months and warn patients to seek medical attention promptly if signs of liver failure occur. Abrupt withdrawal should be avoided because of rebound tachycardia and hypertension. It is typically started at 2 mg daily and increased gradually in increments of 2 mg to a maximum of 36 mg in divided doses (27). A meta-analysis suggested that paradoxical weakness is less of a problem with tizanidine than with baclofen or benzodiazepines (28).
Benzodiazepines
Several benzodiazepines can be used in the management of spasticity, most commonly diazepam and clonazepam. Diazepam has been studied in cross-over and comparison studies in MS, cerebral palsy and paraplegia, with similar efficacy to other drugs. However, the side effects of sedation, respiratory suppression and impairment of attention and memory may limit its use. Tolerance and dependency also frequently occur. Abrupt withdrawal should be avoided as this can precipitate seizures (26). Diazepam can be started at 2 mg at night and increased gradually to a maximum of 60 mg per day in three divided doses. Clonazepam, which has a shorter half-life than diazepam, may also be used, starting at 250-500 µg at night and increased slowly as tolerated to a maximum of 8 mg in three to four divided doses.
Dantrolene sodium
Dantrolene sodium acts directly on skeletal muscle and can therefore be used in conjunction with centrally acting agents with the advantage of causing fewer centrally mediated suppressant side effects, although drowsiness, dizziness, fatigue and respiratory depression are still possible. The drug can also cause severe diarrhea which should prompt withdrawal. A major limiting side effect is the risk of potentially life-threatening hepatotoxicity, usually at doses of greater than 400 mg per day. Liver function tests should be monitored regularly and patients informed about how to recognize signs of liver failure and to seek prompt medical attention in this event. Dantrolene sodium has been studied in stroke, MS, spinal cord injury and cerebral palsy, showing improved measures of spasticity, but again no functional benefit (27). Dantrolene sodium can be started at 25 mg daily and increased on a weekly basis as tolerated to a maximum of 100 mg four times daily. The usual dose is 75 mg three times daily.
Gabapentin
There have been a few small trials in MS and spinal cord injury which have shown good tolerability and beneficial effects on measures of spasticity (27). Gabapentin is also used in the treatment of epilepsy, migraine and neuropathic pain, and so if these are also present, gabapentin may represent a rational treatment choice. Its side effects include gastrointestinal effects, dry mouth, weight gain and dizziness. Abrupt withdrawal may cause anxiety, insomnia and nausea and should therefore be avoided. It is usually commenced at 300 mg per day, either at night or in three divided doses, and can be increased gradually to a maximum of 3.6 g daily in three divided doses. A common maintenance dose is 300 mg three times daily.
Quinine sulphate
Quinine sulphate is used extensively in the elderly for painful nocturnal cramps which are often associated with spasticity. However, the US Food & Drug Administration has issued a safety announcement indicating that quinine sulphate is specifically not approved for the treatment of nocturnal leg cramps because of the risk of serious adverse events including fatalities (29). However, its use continues in other domains, although not commonly in FRDA, perhaps because serious adverse reactions have included cardiac arrhythmias and cardiac conduction defects. Its use is therefore not generally recommended in FRDA.
Other oral medications
Many other medications have shown some evidence of efficacy as possible anti-spasticity agents, including methocarbamol, levitiracetam, lamotrigine, pregabalin, progabide, clonidine, piracetam, vigabatrin, prazepam, cyproheptidine, L-threonine, thymoxamine, orphenadrine and 3,4-diaminopyridine. There are no specific trials in FRDA.
Cannabis extract & cannabinoids
Anecdotally, it is known that patients sometimes gain benefit from the use of cannabis products in reducing pain and spasticity in a variety of neurological conditions. This has not been specifically studied in FRDA. The main active ingredient of the cannabis plant, Cannabis sativa, is Δ9-tetrahydrocannabinol (THC). This is available in a synthetic form as an oral preparation (Dronabinol), or as mixed Cannabis sativa extract (nabiximols) as an oromucosal spray. A further synthetic cannabinoid, nabilone which also has prominent anti-emetic properties, is also available. Previous trials have also been conducted on a further cannabinoid present in Cannabis sativa, cannabidiol (CBD). Side effects have included sedation, dizziness, dry mouth, disorientation and alteration of mood. Cannabinoids should be avoided with coexistent psychotic illness (30).
Extensive studies of Cannabis sativa extract and synthetic cannabinoids (including two small trials of nabilone) have been undertaken largely in MS, but also in stroke, head injury and spinal cord injury. These have largely shown significant reductions in pain, spasms and spasticity. However, by far the largest of these trials (31, 32), which were both randomized, placebo-controlled trials in MS, have failed to show significant reductions in objective markers of spasticity (the Ashworth score) but did show significant reductions in patient-reported measures of spasticity. Further research is required.
Intramuscular botulinum toxin Injection
Intramuscular botulinum toxin injections are the commonest treatment of focal spasticity, although there are no published trials of botulinum toxin injection for spasticity in FRDA.
A review of consensus statements on intramuscular botulinum toxin injection in the management of lower limb spasticity considered 23 trials which included 1047 patients with stroke, MS, trauma and other causes of spasticity (33). Numerous well-controlled, randomized trials showed significant improvement in objective assessments of spasticity such as the Ashworth and Modified Ashworth Scales. Most trials have shown efficacy in improving passive function but few studies have demonstrated improvements in active function. The authors concluded that patients should have detailed assessment by a multidisciplinary team who could identify the main presenting impairments, such as hypertonia, weakness (in particular weakness that will be unmasked by botulinum toxin), loss of range of joint motion or muscle spasm, and could distinguish muscle overactivity from contracture. They felt that treatment was appropriate if improvements could be realistically expected in areas affecting function and participation, such as gait speed, independence in transferring, hygiene and dressing ability; or in reducing impairments such as pain or contractures. They felt that goal-setting in conjunction with the patient was crucial and that the assessment should identify overactive muscles which could be treated in relation to those treatment goals (33).
Botulinum toxin injection has the advantage of engendering highly focal reductions in muscle overactivity without systemic side effects. Side effects include local injection site reactions, pain, tenderness and bruising. Local diffusion of the toxin can cause unwanted local weakness, such as difficulties in speech and swallowing caused by injection into the sternocleidomastoid muscles for cervical dystonia. Smaller muscles are usually more easily treated than large muscles because of the lower dose required to diminish muscle activity. Thus, toe clawing and forced finger flexion may be easier to treat than hip and knee flexion deformities, although functional improvement such as improved seating posture may still be possible with the latter. The clinical effect of botulinum toxin injections begins gradually over 4 to 7 days and causes clinically detectable weakness for 3 to 4 months, with benefit then wearing off gradually. Patients are therefore usually injected at 3- to 4-month intervals, if symptoms recur. Long-term treatment appears to be safe, although efficacy may be reduced by the production of neutralizing antibodies with frequent injections. It is estimated that this occurs in approximately 1 in 200 cases after 1 to 4 injections (34), but the incidence may be as high as 4% for more prolonged treatment. More than 10% of patients may develop ‘botulinum toxin resistance’ after prolonged use, with the greatest incidence occurring in patients receiving larger or more frequent doses (35).
There is evidence that the beneficial effects of botulinum toxin injection are augmented and prolonged by adjunctive therapies at the time or immediately after injection, such as stretching, taping, casting, dynamic orthoses or electrical muscle stimulation (33). It is therefore vital that botulin toxin injections are combined with a timely course of physical therapy.
Alcohol and phenol injection
Local injection of dilute alcohol or phenol with the intention of chemical destruction of a nerve (neurolysis) has been used in the treatment of focal spasticity, but has been largely superseded by botulin toxin injections (27, 36). Neurophysiological guidance is required to identify the target nerve and the procedure can be poorly tolerated, especially in children. There is a significant risk of long-term pain, paraesthesiae and even causalgia, particularly if the targeted nerve contains sensory fibers. There may also be local tissue damage causing edema or venous thrombosis. The most commonly used interventions are tibial nerve block for foot deformities, and obturator nerve block for scissoring gait or with the intention of improving perineal hygiene.
Intrathecal baclofen and phenol
Ben Smail and colleagues (17) described a 39-year old man with FRDA (heterozygous GAA expansion and T317G point mutation) with painful and disabling spasms who was treated successfully with intrathecal baclofen. The patient had disease onset at age 10 years and lost the ability to walk at the age of 33 years. He had frequent painful extensor spasms and less frequent flexor spasms of the lower limbs and back that periodically ejected him from his wheelchair. He required two belts to maintain him in the wheelchair and was unable to sleep for more than half an hour at a time. Frequent tonic activity of soleus and gastrocnemius was measured by electromyography (EMG), which increased after manual brushing of the soles of the feet. Oral baclofen (90 mg) and dantrolene sodium (300 mg) had been ineffective. A dose-dependent decrease in extensor spasms and associated pain was observed with test injections of 50 to 100 µg intrathecal baclofen. Tonic extensor activity of soleus and gastrocnemius began to decrease 30 minutes after intrathecal injection of 100 µg baclofen, and completely disappeared after 55 minutes. Clinically observed spasms and pain were noted to disappear for 6 hours after intrathecal injection of 75 µg baclofen. The intrathecal baclofen was very well tolerated. The patient’s sleep returned to normal and wheelchair positioning was facilitated. He underwent implantation of an intrathecal baclofen-delivering pump, providing a continuous dose amounting to 100 µg daily. After five months he returned to work, having not been able to work for a year.
There are no systematic trials of intrathecal baclofen in FRDA. There are however, well-conducted, placebo-controlled trials in other causes of spasticity showing benefit, particularly in severely disabled individuals with muscle overactivity mainly confined to the lower limbs, such as is commonly seen in FRDA (26). Baclofen is delivered into the intrathecal space via a catheter linked to a programmable pump which is implanted into the abdomen. This has the advantage of requiring far lower concentrations of baclofen than oral medication and causes fewer systemic side effects. However, the procedure is invasive, expensive and requires long-term continuous monitoring from a multidisciplinary team. The pump reservoir periodically requires recharging and the pump battery replacing. Patients must therefore be selected carefully and usually are those with severe spasticity who have failed other physical and pharmacological methods of spasticity control. Potential response to treatment is usually tested by bolus dose delivery of baclofen by lumbar puncture or temporary catheter. This will ensure anti-gravity muscle strength and control is adequate once the spasticity is reduced. Complications include local wound or device infection which may necessitate removal of the system. Failure of the pump or catheter can cause a catastrophic and potentially fatal syndrome of abrupt baclofen withdrawal causing increased spasticity, anxiety, confusion, hallucinations, seizures and hyperthermia (36). The symptoms may be treated with oral baclofen or other anti-spasmodics whilst the team looking after the pump is alerted.
Intrathecal phenol has also been used, but because of the destructive nature of the agent, its use is restricted to those patients with severe lower limb spasticity who cannot be managed by alternative means. Because of the effects on bladder, bowel and sexual function, careful patient selection and informed consent is vital (27).
3.4.3.2 Physical Therapy
There are limited studies specifically investigating the effects of physical therapy on spasticity in individuals with FRDA. Several papers describe general rehabilitation strategies in patients affected with ataxia (37, 38). A fundamental feature of physical therapy management of spasticity is muscle lengthening by passive stretch with the aim of maintaining range and preventing the formation of contractures. This can be achieved temporarily by the physical therapist, and for more prolonged periods by splinting, casting, orthoses, standing, or correct positioning using wedges, cushions and T-rolls. Splinting can involve firm materials or softer supportive substances such as foam, sheepskin or even lycra. However, a Cochrane review of stretch as an intervention for the prevention of contractures in those with neurological conditions at risk of developing contractures found moderate to high quality evidence to indicate that stretch does not have clinically important immediate, short-term or long-term effects on joint mobility (39). There was also little or no effect of stretch on pain, spasticity, activity limitation, participation, restriction or quality of life. Thirty-five studies with 1391 participants met the inclusion criteria, but no study performed stretch for more than seven months. The authors could therefore not comment on the effects of stretch performed for longer than seven months. In contrast to this 2010 review, Paleg and Livingstone (40) found standing machine stretches for 30 minutes, five days per week improved range of movement and activity for individuals with stroke and spinal cord injury, but there was mixed evidence for other neurological populations (again there were no participants with FRDA).
Good quality, well-fitted orthoses prepared in a specialist orthotics department are also important, if indicated, in the management of spasticity. Promoting the strength of muscles by active exercise which antagonize the overactive spastic muscle is also important in reducing spasticity. Again, correct positioning may optimize the functioning of muscle groups which can work to counteract spasticity. Weights and resistant devices can be used to promote muscle strength. Active exercise is generally more effective than passive exercise, if the patient is able, and the consequent increased cardiovascular fitness helps to combat fatigue and allows further exercise. Repeated practicing is used to reinforce motor learning which improves function. If one limb is more profoundly affected than the other, it may be beneficial to constrain the better limb, forcing the weaker limb to undertake more work, and thus strengthen it. Conversely, compensatory techniques may be taught to allow the patient to function optimally. There is some evidence for electrical stimulatory devices in the treatment of spasticity, including the Functional Electrical Stimulator, the Foot Drop Stimulator and the Transcutaneous Nerve Stimulator (27, 41).
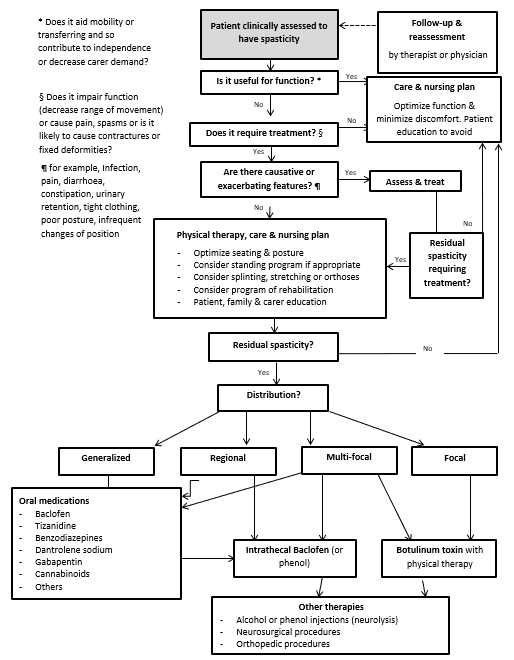
Physical therapists have a critical role to play in educating patients and caregivers as to correct posture, muscle use and the identification and avoidance of features which aggravate spasticity, such as tight clothing, poorly adjusted wheelchairs, pain and infection. Physical therapy has to become ‘a way of life’ for the patient. Therapists should also promote motivation, positive thinking and psychological support (see Figure 3.4.1).
Figure 3.4.1: Algorithm for management of spasticity – modified from Thompson et al (25) and Olver et al (33)
3.4.3.3 Surgical Treatment
Surgical treatments are generally considered options of last resort when physical and pharmacological interventions have failed. However, in exceptional cases, particularly in the case of a fixed, non-reducible deformity, they may be considered.
Orthopedic procedures
If severe hypertonia results in established limb deformities such as ankle equinovarus, orthopedic procedures such as tendon lengthening, tenotomy or tendon transfer may be indicated. Delatycki and colleagues (42) studied 32 patients with FRDA and equinovarus deformity of the foot. They were assigned to three groups: mild, requiring splinting alone (n=15); moderate but reducible, requiring botulinum toxin injection (n=8); and severe and not fully reducible, requiring surgery (n=9). Seven patients ultimately went on to have surgery involving lengthening of the Achilles tendon to correct the equinus deformity, and transfer of the tibialis posterior tendon to peroneus brevis to correct the varus deformity. None of these seven patients was able to stand to transfer independently before surgery, whereas all seven were able to do this post-operatively. None were able to walk independently after surgery. Transfers and mobility subscales of the Modified Barthel Index and the Functional Independence Measure showed significant change between the pre- and post-operative periods. Three patients had significant surgical complications (two cases of aspiration and one of pulmonary embolus requiring plasminogen activator) but each recovered fully. The authors recommended aggressive management of foot deformities.
Neurosurgical procedures
With the exception of intrathecal baclofen pump insertion, the neurosurgical treatment of spasticity is only considered if all other medical and physical methods have failed. Procedures include peripheral neurotomies, dorsal rhizotomies and microsurgical ablation of the dorsal root entry zone (DREZotomy). It is mainly indicated when patients have focal spasticity without useful mobility, and ablation of specific nerve fibers will result in improvement in a specific focal symptom, such as tibial nerve ablation for equinovarus deformity of the foot. It is therefore vital to have clear functional goals, such as decreased pain, improved comfort, or improved posture or transferring. All these methods are now very rare as the techniques are destructive and irreversible, and the results variable. Inserted neurostimulators of the cervical spinal cord and cerebellar cortex were also previously used, largely in cerebral palsy. No trials have been conducted in FRDA (43).
Strengthening and stretching with or without pharmacological therapy
QUESTION: Should strengthening and stretching (including standing machine exercises and serial casting) with or without pharmacological therapy versus pharmacological therapy only or no intervention be used for people with spasticity with Friedreich ataxia?

LEVEL OF EVIDENCE: ⨁⨁◯◯
RECOMMENDATION: We suggest non-pharmacological (physiotherapy) treatment, such as strengthening and stretching, should be used as a first option in the management of spasticity (and its secondary consequences such as mobility decline), prior to considering pharmacological interventions for individuals with Friedreich ataxia with spasticity.
We suggest physiotherapy/rehabilitation interventions (such as strengthening, stretching, serial casting and standing machine stretch) are used to enhance the effects of pharmacological therapy for management of spasticity in individuals with Friedreich ataxia. This should occur when non-pharmacological treatment alone does not address the individual’s problems and/or treatment goals.
JUSTIFICATION: Although there are no studies that have directly examined stretching and strengthening for the management of spasticity in individuals with Friedreich ataxia, it is common practice to trial more conservative therapies prior to initiating pharmacological treatment.
There are no published studies in Friedreich ataxia; however clinical expert observations in clinical practice suggests that physiotherapy interventions as an adjunct to pharmacotherapy is a beneficial approach in managing spasticity. Similarly, a 2014 systematic review (44) suggests this approach may be slightly more effective than pharmacotherapy management alone.
Clinicians noted there were regional differences in the approach to supplement pharmacotherapy with physiotherapy interventions. A case by case review was recommended to ensure the appropriate therapy was provided to each individual. Use of adjunctive physiotherapy usually comes after a stretching intervention.
SUBGROUP CONSIDERATION: This recommendation is for individuals with Friedreich ataxia with spasticity.
Evidence to Recommendation Table PDFLocal pharmacological therapy
QUESTION: Should local pharmacotherapy versus no intervention be used for people who have focal spasticity and this is impacting on function or pain with Friedreich ataxia?
LEVEL OF EVIDENCE: ⨁◯◯◯
RECOMMENDATION: We suggest local pharmacological (i.e. injection of botulinum toxin) management of spasticity for individuals with Friedreich ataxia over no local spasticity management in the following circumstances: a thorough assessment is conducted to weigh up the negative and positive effects of this intervention; spasticity is significantly affecting mobility, function, pain or positioning, and conservative treatment options (such as physiotherapy) are no longer effective.
JUSTIFICATION: There are no published studies in Friedreich ataxia; however clinical expert observations in clinical practice suggests this may a beneficial approach. Expert opinion suggests pharmacological intervention should be coupled with physiotherapy interventions (refer to other recommendations in this topic). This recommendation is supported by a similar recommendation from the Ataxia UK Medical Guidelines (45).
SUBGROUP CONSIDERATION: This treatment for spasticity may pose greater risks to mobility in ambulant individuals as compared to individuals who are no longer ambulant. However, this is not substantiated with published evidence.
Evidence to Recommendation Table PDFSystemic pharmacological therapy (baclofen, tizanidine, etc)
QUESTION: Should systemic pharmacotherapy (baclofen, tizanidine, gabapentin, dantrolene sodium, benzodiazepines, other) versus no intervention be used for people with spasticity and spasms/cramps with Friedreich ataxia?
LEVEL OF EVIDENCE: ⨁◯◯◯
RECOMMENDATION: We conditionally recommend offering systemic pharmacotherapy (baclofen, tizanidine, gabapentin, dantrolene sodium, benzodiazepines, other) for the management of generalized spasticity and spasms in individuals with Friedreich ataxia, with a view to reducing the severity of spasticity and the frequency of spasms and cramps, which may improve mobility and upper limb function and reduce pain.
JUSTIFICATION: The studies on systematic pharmacological treatment for spasticity and spasm in Friedreich ataxia are all small and only oral or intrathecal baclofen has been studied. Most evidence is derived from studies of spasticity in multiple sclerosis which show evidence of efficacy, although with varying quality of evidence. However, clinical expert observations in clinical practice suggests this can be a beneficial approach in Friedreich ataxia.
SUBGROUP CONSIDERATION: This recommendation is for individuals with Friedreich ataxia with spasticity and spasms/cramps.
Evidence to Recommendation Table PDFSystemic pharmacological therapy (non-licensed: cannabis, other)
QUESTION: Should systemic pharmacotherapy (non-licensed: cannabis, other) versus no intervention be used for people with spasticity and spasms/cramps with Friedreich ataxia?
LEVEL OF EVIDENCE: ⨁◯◯◯
RECOMMENDATION: We cannot recommend either the use or non-use of systemic pharmacotherapy (non-licensed: cannabis, other) to manage spasticity and spasms/cramps in people with Friedreich ataxia.
JUSTIFICATION: There is no published evidence related to the use or non-use of non-licensed systemic pharmacotherapy for individuals with Friedreich ataxia. The clinical experience of the authors indicated patients reported deteriorating function and mobility with self-prescribed administration. Conversely, anecdotal information relayed to the expert authors report efficacy in individuals who have been resistant to other treatments, with associated reduction in anxiety and improvement in sleep.
SUBGROUP CONSIDERATION: Although there is no conclusive evidence, clinical experience suggests this intervention may be more beneficial for individuals with treatment-resistant troubling spasticity and spasm.
Evidence to Recommendation Table PDFNeuromodulation
QUESTION: Should neuromodulation (broad – TMS, other) versus no intervention be used for people with spasticity with Friedreich ataxia?
LEVEL OF EVIDENCE: ⨁◯◯◯
RECOMMENDATION: We conditionally recommend against the use of neuromodulation for the treatment of spasticity in individuals with Friedreich ataxia.
JUSTIFICATION: There is no published evidence of the effects of neuromodulation for spasticity management in individuals with Friedreich ataxia. Benussi and colleagues (46) reviewed 10 published studies (n=116) which confirmed the favorable effect of tDCS on a range of motor domains including gait, balance and upper limb function in neurodegenerative ataxias, but did not report effects on spasticity.
A review specifically examining the effect of repetitive transcranial magnetic stimulation or transcranial direct current stimulation on spasticity of the limbs found 30 randomized controlled trials, with the majority of the studies including participants post-stroke or with multiple sclerosis (47). The findings suggest neuromodulation is useful in reducing spasticity, but its effects depend on the applied hemisphere and the underlying pathology. Furthermore, they recommended that neuromodulation should be applied as a pre-curser to other medical and/or physical therapy.
SUBGROUP CONSIDERATION: This recommendation is for individuals with Friedreich ataxia with spasticity.
Evidence to Recommendation Table PDFAggravating factors such as infection, pain, constipation, diarrhea, dehydration and pressure sores should be considered and treated in the context of acute onset or exacerbation of spasticity and/or ataxia.
Individuals with Friedreich ataxia, families and caregivers should be educated to monitor the development of spasticity and incipient contractures.
On implementation of an anti-spasticity intervention, individuals with Friedreich ataxia may benefit from reassessment as the treatment of spasticity can unmask weakness and cause deterioration in gait and standing transfers. Individuals should be warned of this phenomenon before anti-spasticity interventions are commenced.
Lay summary of clinical recommendations for spasticity and spasms in Friedreich ataxia
Spasticity is where muscles remain contracted or contract involuntarily and become stiff or tight. This prevents normal hand or leg movement and can impact on normal walking pattern. Spasms are a form of spasticity where the muscle forcibly contracts when completing an unrelated activity, such as sneezing or re-positioning in a chair. Spasticity or spasm may or may not cause pain in the muscles.
In people with Friedreich ataxia, spasticity can be present in a single muscle or in many muscles. Published research shows that spasticity can be present in the legs or hands in individuals with Friedreich ataxia.
Why these recommendations?
There is limited published research on the treatment of spasticity in Friedreich ataxia; however, some of the research in other conditions, such as multiple sclerosis, can be carefully applied to manage spasticity in people with Friedreich ataxia. Deciding on the best treatment depends on how severe the spasticity is, where it occurs and how it impacts on function and/or pain. A thorough assessment with your neurologist and/or physiotherapist will help to work out the best approach.
These recommendations suggest firstly managing spasticity with physiotherapy treatment, such as stretching and strengthening. However, if this no longer provides any benefit, then a number of different medications can be considered for each individual to manage their spasticity. After a thorough assessment of spasticity and its impact on pain, posture and ability to physically function:
1) if spasticity is present in a few select muscles, we suggest the use of an injection of botulinum neurotoxin rather than no treatment at all.
2) if spasticity in present in a number of muscles, we suggest the use of medication, taken orally, rather than no treatment at all. These medications may include baclofen, gabapentin, clonidine, clonazepam.
3) If neither of the above options are helpful, we recommend consideration of intrathecal (into the spinal cord) baclofen or phenol injections.
This recommendation suggests that physiotherapy treatment may enhance the benefits of these medications.
With no evidence published for its use in the treatment of spasticity in people with Friedreich ataxia, neuromodulation (stimulation of the brain) or non-licenced medications such as cannabis cannot be recommended.
What does this mean for you as a person living with Friedreich ataxia or caring for someone living with Friedreich ataxia?
We suggest that physiotherapy should be your first treatment for spasticity and spasm. If that does not help, it is important for you to discuss the benefits and possible side effects of medications for spasticity and monitor and report any changes in your physical function with your doctor, neurologist and/or physiotherapist.
Who are these recommendations specifically for?
These recommendations are for all individuals with Friedreich ataxia who have spasticity and/or spasms.
Professor, Queen Square Institute of Neurology, UCL, London, UK
Email: p.giunti@ucl.ac.uk
Sarah C. Milne, BPhysio, PhD
Physiotherapist, Murdoch Children’s Research Institute, Melbourne, Victoria, Australia
Email: sarah.milne@mcri.edu.au
Michael H. Parkinson, MBBS, PhD
Consultant Neurologist, National Hospital for Neurology & Neurosurgery, London, UK
Sub H. Subramony, MD
Professor of Neurology and Pediatrics, University of Florida College of Medicine, Gainesville, Florida, USA
Email: s.subramony@neurology.ufl.edu
2. Bhidayasiri R, Perlman SL, Pulst SM, Geschwind DH. Late-onset Friedreich ataxia: phenotypic analysis, magnetic resonance imaging findings, and review of the literature. Arch Neurol. 2005;62(12):1865-9.
3. Geoffroy G, Barbeau A, Breton G, Lemieux B, Aube M, Leger C, et al. Clinical description and roentgenologic evaluation of patients with Friedreich’s ataxia. Can J Neurol Sci. 1976;3(4):279-86.
4. Harding AE. Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981;104(3):589-620.
5. Montermini L, Andermann E, Labuda M, Richter A, Pandolfo M, Cavalcanti F, et al. The Friedreich ataxia GAA triplet repeat: premutation and normal alleles. Hum Mol Genet. 1997;6(8):1261-6.
6. Badhwar A, Jansen A, Andermann F, Pandolfo M, Andermann E. Striking intrafamilial phenotypic variability and spastic paraplegia in the presence of similar homozygous expansions of the FRDA1 gene. Mov Disord. 2004;19(12):1424-31.
7. Milne SC, Corben LA, Yiu E, Delatycki MB, Georgiou-Karistianis N. Gastrocnemius and soleus spasticity and muscle length in Friedreich’s ataxia. J Clin Neurosci. 2016;29:29-34.
8. Corben LA, Yiu EM, Tai G, Milne SC, Lynch B, Delatycki MB. Probing the multifactorial source of hand dysfunction in Friedreich ataxia. J Clin Neurosci. 2019;64:71-6.
9. Ragno M, De Michele G, Cavalcanti F, Pianese L, Monticelli A, Curatola L, et al. Broadened Friedreich’s ataxia phenotype after gene cloning. Minimal GAA expansion causes late-onset spastic ataxia. Neurology. 1997;49(6):1617-20.
10. Gates PC, Paris D, Forrest SM, Williamson R, Gardner RJ. Friedreich’s ataxia presenting as adult-onset spastic paraparesis. Neurogenetics. 1998;1(4):297-9.
11. Coppola G, De Michele G, Cavalcanti F, Pianese L, Perretti A, Santoro L, et al. Why do some Friedreich’s ataxia patients retain tendon reflexes? A clinical, neurophysiological and molecular study. J Neurol. 1999;246(5):353-7.
12. Richter A, Poirier J, Mercier J, Julien D, Morgan K, Roy M, et al. Friedreich ataxia in Acadian families from eastern Canada: clinical diversity with conserved haplotypes. Am J Med Genet. 1996;64(4):594-601.
13. Barbeau A. The Quebec Cooperative Study of Friedreich’s Ataxia: 1974-1984–10 years of research. Can J Neurol Sci. 1984;11(4 Suppl):646-60.
14. Gellera C, Castellotti B, Mariotti C, Mineri R, Seveso V, Didonato S, et al. Frataxin gene point mutations in Italian Friedreich ataxia patients. Neurogenetics. 2007;8(4):289-99.
15. Cossée M, Dürr A, Schmitt M, Dahl N, Trouillas P, Allinson P, et al. Friedreich’s ataxia: point mutations and clinical presentation of compound heterozygotes. Ann Neurol. 1999;45(2):200-6.
16. Corben LA, Yiu EM, Tai G, Milne SC, Lynch B, Delatycki MB. Probing the multifactorial source of hand dysfunction in Friedreich ataxia. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2019;64:71-6.
17. Ben Smail D, Jacq C, Denys P, Bussel B. Intrathecal baclofen in the treatment of painful, disabling spasms in Friedreich’s ataxia. Mov Disord. 2005;20(6):758-9.
18. National Clinical Guideline Centre (UK). Multiple Sclerosis: Management of Multiple Sclerosis in Primary and Secondary Care. London: National Institute for Health and Care Excellence (UK); 2014 [Available from: https://www.ncbi.nlm.nih.gov/books/NBK328181/.
19. Shakespeare DT, Boggild M, Young C. Anti-spasticity agents for multiple sclerosis. Cochrane Database Syst Rev. 2003(4):CD001332.
20. Amatya B, Khan F, La Mantia L, Demetrios M, Wade DT. Non pharmacological interventions for spasticity in multiple sclerosis. Cochrane Database Syst Rev. 2013(2):CD009974.
21. Taricco M, Adone R, Pagliacci C, Telaro E. Pharmacological interventions for spasticity following spinal cord injury. Cochrane Database Syst Rev. 2000(2):CD001131.
22. Ashworth NL, Satkunam LE, Deforge D. Treatment for spasticity in amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2012(2):CD004156.
23. Ade-Hall RA, Moore AP. Botulinum toxin type A in the treatment of lower limb spasticity in cerebral palsy. Cochrane Database Syst Rev. 2000(2):CD001408.
24. Hoare BJ, Wallen MA, Imms C, Villanueva E, Rawicki HB, Carey L. Botulinum toxin A as an adjunct to treatment in the management of the upper limb in children with spastic cerebral palsy (UPDATE). Cochrane Database Syst Rev. 2010(1):CD003469.
25. Thompson AJ, Jarrett L, Lockley L, Marsden J, Stevenson VL. Clinical management of spasticity. J Neurol Neurosurg Psychiatry. 2005;76(4):459-63.
26. Abbruzzese G. The medical management of spasticity. Eur J Neurol. 2002;9 Suppl 1:30-4; discussion 53-61.
27. Stevenson VL. Rehabilitation in practice: Spasticity management. Clin Rehabil. 2010;24(4):293-304.
28. Groves L, Shellenberger MK, Davis CS. Tizanidine treatment of spasticity: a meta-analysis of controlled, double-blind, comparative studies with baclofen and diazepam. Adv Ther. 1998;15(4):241-51.
29. US Food and Drug Administration. New risk management plan and patient Medication Guide for Qualaquin (quinine sulfate) 2010 [Available from: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/fda-drug-safety-communication-new-risk-management-plan-and-patient-medication-guide-qualaquin.
30. Karst M, Wippermann S, Ahrens J. Role of cannabinoids in the treatment of pain and (painful) spasticity. Drugs. 2010;70(18):2409-38.
31. Collin C, Davies P, Mutiboko IK, Ratcliffe S, Sativex Spasticity in MSSG. Randomized controlled trial of cannabis-based medicine in spasticity caused by multiple sclerosis. Eur J Neurol. 2007;14(3):290-6.
32. Zajicek J, Fox P, Sanders H, Wright D, Vickery J, Nunn A, et al. Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): multicentre randomised placebo-controlled trial. Lancet. 2003;362(9395):1517-26.
33. Olver J, Esquenazi A, Fung VS, Singer BJ, Ward AB, Cerebral Palsy I. Botulinum toxin assessment, intervention and aftercare for lower limb disorders of movement and muscle tone in adults: international consensus statement. Eur J Neurol. 2010;17 Suppl 2:57-73.
34. Yablon SA, Brashear A, Gordon MF, Elovic EP, Turkel CC, Daggett S, et al. Formation of neutralizing antibodies in patients receiving botulinum toxin type A for treatment of poststroke spasticity: a pooled-data analysis of three clinical trials. Clin Ther. 2007;29(4):683-90.
35. Greene P, Fahn S, Diamond B. Development of resistance to botulinum toxin type A in patients with torticollis. Mov Disord. 1994;9(2):213-7.
36. Tilton A, Vargus-Adams J, Delgado MR. Pharmacologic treatment of spasticity in children. Semin Pediatr Neurol. 2010;17(4):261-7.
37. Milne SC, Corben LA, Georgiou-Karistianis N, Delatycki MB, Yiu EM. Rehabilitation for individuals with genetic degenerative ataxia: A systematic review. Neurorehabil Neural Repair. 2017;31(7):609-22.
38. Rodriguez-Diaz JC, Velazquez-Perez L, Rodriguez Labrada R, Aguilera Rodriguez R, Laffita Perez D, Canales Ochoa N, et al. Neurorehabilitation therapy in spinocerebellar ataxia type 2: A 24-week, rater-blinded, randomized, controlled trial. Mov Disord. 2018;33(9):1481-7.
39. Katalinic OM, Harvey LA, Herbert RD, Moseley AM, Lannin NA, Schurr K. Stretch for the treatment and prevention of contractures. Cochrane Database Syst Rev. 2010(9):CD007455.
40. Paleg G, Livingstone R. Systematic review and clinical recommendations for dosage of supported home-based standing programs for adults with stroke, spinal cord injury and other neurological conditions. BMC Musculoskelet Disord. 2015;16:358.
41. Richardson D. Physical therapy in spasticity. Eur J Neurol. 2002;9 Suppl 1:17-22; dicussion 53-61.
42. Delatycki MB, Holian A, Corben L, Rawicki HB, Blackburn C, Hoare B, et al. Surgery for equinovarus deformity in Friedreich’s ataxia improves mobility and independence. Clin Orthop Relat Res. 2005;430:138-41.
43. Lazorthes Y, Sol JC, Sallerin B, Verdie JC. The surgical management of spasticity. Eur J Neurol. 2002;9 Suppl 1:35-41; dicussion 53-61.
44. Kinnear BZ, Lannin NA, Cusick A, Harvey LA, Rawicki B. Rehabilitation therapies after botulinum toxin-A injection to manage limb spasticity: a systematic review. Phys Ther. 2014;94(11):1569-81.
45. de Silva RN, Greenfield J, Cook A, Bonney H, Vallortigara J, Hunt B, et al. Guidelines on the diagnosis and management of progressive ataxia in adults. Orphanet J Rare Dis. 2019;14:51.
46. Benussi A, Pascual-Leone A, Borroni B. Non-invasive cerebellar stimulation in neurodegenerative ataxia: A literature review. Int J Mol Sci. 2020;21(6).
47. Leo A, Naro A, Molonia F, Tomasello P, Saccà I, Bramanti A, et al. Spasticity management: The current state of transcranial neuromodulation. PM R. 2017;9(10):1020-9.
These Guidelines are systematically developed evidence statements incorporating data from a comprehensive literature review of the most recent studies available (up to the Guidelines submission date) and reviewed according to the Grading of Recommendations, Assessment Development and Evaluations (GRADE) framework © The Grade Working Group.
This chapter of the Clinical Management Guidelines for Friedreich Ataxia and the recommendations and best practice statements contained herein were endorsed by the authors and the Friedreich Ataxia Guidelines Panel in 2022.
It is our expectation that going forward individual topics can be updated in real-time in response to new evidence versus a re-evaluation and update of all topics simultaneously.